成功通過 MDSAP 稽核的關鍵考量
專文
我們的專家向醫療器材製造商深入解析準備 MDSAP 稽核的流程。閱讀本文,了解最佳做法,確保符合法規要求。
「醫療器材單一稽核計畫」 (Medical Device Single Audit Program, MDSAP) 納入了 ISO 13485:2016 的要求,並涵蓋 5 個國家的醫療器材法規,分別為澳洲、巴西、加拿大、日本及美國。我們的 MDSAP 服務介紹網頁詳細說明了獲得 MDSAP 證書的優勢。現在透過本專文,我們將探討獲得 MDSAP 證書之前需要完成的重要步驟。
為了獲得 MDSAP 認證,製造商的品質管理系統 (Quality Management System, QMS) 必須符合 ISO 13485:2016 標準的所有適用要求。此外,系統還須結合/整合醫療器材法規,以及製造商正在或打算販售產品之該 MDSAP 會員國的要求。
即便 MDSAP 涵蓋五大會員國家,但 QMS 為僅需符合製造商在認證當下所相應的國家要求。因此,若製造商最初只有計畫將相關器材銷往美國與澳洲,則其 QMS 在進行首次註冊稽核時只需符合美國食品藥物管理局 (U.S. Food and Drug Administration, FDA) 及澳洲藥品管理局 (Therapeutic Goods Administration, TGA) 的適用規範。而假若未來有進入其他 MDSAP 國家市場的計畫,QMS 即需隨之調整,以符合額外國家的法規要求,並可相應擴展證書適用範圍。
概括而言,認證流程包含四個步驟:
- 稽核前 (Pre-audit) 階段:報價、合約簽訂與稽核排程
- 第 1 階段 (Stage 1) 與第 2 階段 (Stage 2) 稽核
- 稽核後 (Post-audit) 活動
- 證書 (Certification) 簽發
製造商需要充分準備,且展現強烈的管理層承諾與行動,才能成功完成每個步驟。![]()
稽核前階段
在簽訂合約之前,客戶需要提供基本資訊,包括組織與設施概況、想要銷售的醫療器材產品、員工數及其他細節。UL Solutions 將根據初步資訊提供報價,並在合約簽回前解答與報價相關的問題,包括首次註冊稽核的預計時程。若客戶接受報價,客戶與稽核機構 (auditing organization, AO) 將簽訂合約,並安排註冊稽核的時間。
第 1 階段稽核
第 1 階段稽核 (S1) 的主要目的是評估客戶的 QMS 是否已準備好接受 MDSAP 認證。這個階段所關注的重點將會是可明確定義 QMS 的文件化流程 (如 SOP 及相關文件)。SOP 應涵蓋 ISO 13485:2016 及 MDSAP 稽核應對方法文件 (MDSAP AU P0002) 中所定下的所有適用流程。這份 MDSAP 稽核應對方法文件可在 MDSAP 文件網站公開取得。
QMS 應該要能可以反映對 ISO 13485:2016 標準及 MDSAP 稽核應對方法文件指引的完整理解。
可選擇要在現場或遠端進行第 1 階段稽核,我們建議最好能夠將所有 SOP 做電子格式的存檔,以便遠端檢閱。此外,正由於稽核機構 (AO) 的品質手冊通常都是所有 QMS 的核心項目,故其將成為這階稽核的重點審查。
需要注意的是,在部分 MDSAP 會員國,境外製造商必須尋求當地機構協助辦理初步市場授權,但可能在其他國家,這個部分則是可選要求。
- 在澳洲,境外製造商必須找到澳洲當地,予以代表向 TGA 提出申請註冊其醫療器材。這個代表稱為「澳洲贊助者」(Australian sponsor)。
- 在巴西,境外製造商必須與當地機構合作,該機構將成為巴西註冊持有者 (Brazilian registration holder, BRH),以負責獲得在巴西的行銷授權。
- 在日本銷售醫療設備,製造商必須委任當地代表 ── 稱之行銷授權持有者 (marketing authorization holder, MAH),以代表製造商與厚生勞動省 (Ministry of Health, Labour and Welfare, MHLW) 溝通。
- 在加拿大,若欲銷售第 2 類 (Class 2) 及以上類別的醫療器材,製造商必須持有 MDSAP 證書。在 MDSAP 五個會員國中,目前僅加拿大對 MDSAP 認證做出強制性要求。但若販售之產品為加拿大衛生部 (Health Canada, HC) 指定的第 1 類醫材,就不需要 MDSAP 證書。在加拿大,境外製造商可選擇與加拿大境內的進口商合作,但這並非強制要求。製造商亦可選擇直接向加拿大衛生部申請醫療器材許可,或選擇尋求第三方法規顧問協助與衛生部的溝通,但這些也不是強制要求。
- 在美國,醫療器材製造商可直接與美國 FDA 溝通。
在 S1 稽核期間,每一家相對應的法規代理機構必須要顯示對基本要求的了解。若 S1 稽核中發現任何不足,這些缺失將會反饋到其客戶端,並必須在第 2 階段稽核 (S2) 前完成改善。根據缺失程度,可能會導致兩種結果:
- 認定客戶 QMS 未達到 MDSAP 要求,並可能建議進行新的 S1 稽核;或者
- 認定缺失的性質相對較為輕微,因此下一步仍可走到 S2 稽核階段。不過取決於缺失的嚴重程度,S2 稽核時間的安排可能會落在 S1 稽核後的 1 到 6 個月內。但若缺失超過六個月仍未改善,則需重新進行 S1 稽核。
第 2 階段稽核
第 2 階段的 MDSAP 稽核 (S2 稽核) 為現場稽核,通常完成需要數日的時間。稽核所需的天數將會在簽約前讓製造商知道。此階稽核的進行是依據 MDSAP 稽核應對方法的模型,並依照以下結構來評估 QMS 的各個方面:
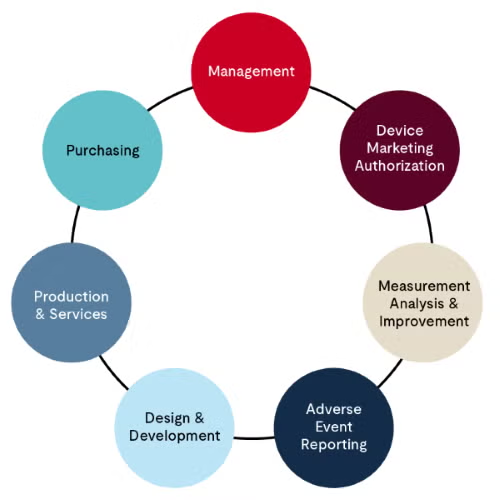
- 管理流程 (Management process) ── 共分為 11 個稽核項目,聚焦在 QMS 整體架構的概覽,包括但不限於:組織架構、管理層承諾、管理審查、文件控制、人力資源與能力評估等。品質手冊將列為評估的核心。
- 醫療器材行銷授權流程 (Device marketing authorization process) ── 此部分著重於在相關法規機構註冊設施以及醫療器材許可程序,可分為 3 個稽核項目。這些流程通常由組織內負責 MDSAP 認證的法規專家管理。
- 量測、分析與改進 (Measurement, analysis, and improvement) ── 顧名思義,這個部分幾乎所有的關注會在 QMS 運作的監測與改進計畫,其中包含客訴處理、CAPA、內部稽核流程、不合格材料與製程管理,以及上市後監控。以上所述的內容皆為核心重點。這些流程通常會由組織內負責 MDSAP 認證的 QA 經理管理。
- 不良事件通報、產品召回與顧問通知 (Adverse event reporting, recalls, and advisory notices) ── 包含了 3 個稽核項目,並依據 MDSAP 稽核應對方法進行評估。這些流程通常由組織內負責 MDSAP 認證的法規事務團隊管理。
- 設計與開發流程 (Design and development process) ── 在 MDSAP 稽核應對方法中,此部分涉及 16 個稽核項目,涵蓋設計與開發新器材產品時的從開始規劃、設計轉移至製造的完整過程,並包含設計變更管理。
- 生產與服務控制流程 (Production and services control process) ── 在 MDSAP 稽核應對方法中,針對生產與服務控制流程,建議涵蓋 29 個稽核項目。這裡涉及醫療器材在製造時會涉及的各個環節,包括但不限於:基礎設施、工作環境、生產製程驗證、生產品質控制測試、倉儲、產品識別與追溯、不合格材料處理及銷售。
稽核後的活動
稽核後的活動可包含兩大重點:
- 製造商須針對稽核期間所找到的不符合項目進行修正;以及
- 稽核機構將會對其稽核結果進行內部法規審查。
在 S1 稽核發現的不符合項目,必須在 S2 稽核時完成改善、消除並驗證。若 S2 稽核仍發現有不符合項目,即會根據 MDSAP 要求,依 1 至 5 級制予以分級。MDSAP 所採用的分級機制基本概念源自於 GHTF/SG3/N19:2012。由於該 GHTF 文件早就在 ISO 13485:2016 發布,因此文件內仍是參照 ISO 13485:2003 標準。然而,該分級機制的原則與概念仍然適用於 MDSAP。若不符合項目被評為第 4 級或第 5 級,或被歸類為 ISO 13485「重大缺失」,則必須在 MDSAP 證書簽發前完成修正。
稽核團隊會將稽核結果提交給內部法規審核團隊。而根據審核結果,證書核發的決定者即會判定該次稽核是否達到簽發 MDSAP 證書的標準,並將最終決定通知製造商。此外,最終稽核報告也會依據 MDSAP 政策提交至 MDSAP REPS 資料庫。
為何選擇 UL Solutions 進行 MDSAP 認證?
|
本文作者
Chira Deka 是 UL Solutions 的 MDSAP 計畫專案經理,並兼任地區主管審核員、證書決定者,以及 ISO 13485、ISO 9001 和 MDSAP 認證計畫的首席稽核員。此外,他亦是 MDD 與 IVDD 計畫的首席稽核員,以及 UKCA 認證計畫的稽核員。Chira 同時還是 IVD 領域的技術專家,並擁有多項相關專利。其曾擔任 NIH SBIR 技術審查小組 (微生物學) 成員,並曾獲得 NIH 及美國商務部 ATP 的研究資助,專注於 IVD 及生物技術領域。
相關服務探索
更多資源下載
- 白皮書 | 醫療器材的 INMETRO 認證